
天使综合征诊疗指南
概述
Angelman氏症候群(天使综合征)(Angelman syndrome,AS)是一种由于母源15q11-13染色体区域的*UBE3A*基因表达异常或功能缺陷引发的神经发育障碍性疾病。主要表现为精神发育迟滞或智力低下,语言、运动或平衡发育障碍,快乐行为(如频繁发笑、微笑或兴奋),小头畸形,癫痫等。
病因和流行病学
母源*UBE3A*基因的表达或功能缺陷导致AS。已明确的分子遗传机制有4种:母源15q11-13缺失、父源15号染色体存在单亲二体(Uniparental disomy, UPD)、母源15q11.2-q13印记缺陷(imprinting defect, ID)和母源*UBE3A*基因发生致病突变。四种分子机制都导致了母源*UBE3A*基因的功能丧失。其中母源15q11~13缺失最常见,多数约长5~7 Mb。
*UBE3A*基因编码的泛素蛋白连接酶E3A参与了泛素化途径,对特定蛋白进行降解。在人类胎儿脑组织和成人额叶皮质中,*UBE3A*基因主要表现为母源表达,父源因甲基化不表达。正常情况下,神经系统由功能性泛素-蛋白酶体系统进行平衡或维持,当*UBE3A*失功能时则可能影响该系统,引起患者黑质、纹状体、海马及小脑浦肯野细胞蛋白泛素化异常。另外,泛素-蛋白酶体系统对细胞功能也至关重要,包括信号转导、细胞周期进程、DNA修复和转录调节。然而,该疾病导致泛素化异常的具体病理生理机制尚不完全清楚。
AS在欧美人群患病率为1/24 000~1/12 000。我国多为散发报道,尚无相关流行病学调查报告。
临床表现
患者在新生儿期常无异常表现。首发症状是6月龄左右出现发育迟滞表现,但典型临床表现多在1岁后出现,临床确定诊断往往需要数年时间。按照临床表现出现频率不同分为:均出现的表现(100%患者)、经常性表现(80%以上患者)、相关性表现(20%~80%患者)3类。
均出现的表现 正常孕产史和出生头围;无出生缺陷和生化指标异常;颅脑MRI/CT除轻微皮质萎缩、髓鞘发育不良外,多无结构异常;运动里程碑落后而无倒退;6~12个月出现严重发育迟缓;语言障碍显示为无或极少量词汇,重复性语言和非语言交往能力强于表达性语言能力;运动或平衡障碍常表现为共济失调以及四肢震颤;异常的行为特征表现为频繁大笑或微笑、明显的兴奋动作或快乐举止、常伴拍手或多动。
经常性表现 头围增长落后,随访至2岁仍表现为小头畸形;3岁前常出现癫痫;异常脑电图:特征性高波幅棘-慢波。
相关性表现 枕部扁平;喜吐舌;吸吮或吞咽障碍;婴儿期喂养困难;肌张力低;巨大下颌;牙间隙宽;频繁流涎;过度咀嚼动作;斜视;皮肤色素减退,与家人相比头发和眼睛颜色浅(仅见于缺失型);下肢腱反射亢进;上举或弯曲上肢,尤其是行走时;步基宽;对热敏感性高;异常睡眠-觉醒周期、睡眠少;迷恋水、纸、塑料;进食相关行为异常;肥胖(多见于年长、非缺失型患者);脊柱侧凸;便秘。
辅助检查
1.遗传学检查 遗传学检查为该病确诊手段。具体包括DNA甲基化分析,这类方法包括甲基化多重连接依赖式探针扩增技术(MS-MLPA)、甲基化PCR(MS-PCR)等;*UBE3A*基因序列分析;SNP-array芯片分析可以检测15号染色体的微缺失和UPD。
2.脑电图 AS具有相对特征性的EEG异常,常能在临床症状明显前及基因诊断前提示本病,从而有助于早期诊断。目前比较公认的AS特征性异常EEG包括3种图形:①δ图形;②θ图形;③后头部棘慢波图形。
诊断
符合AS临床诊断标准共识和(或)分子遗传学检测结果表明母源*UBE3A*等位基因存在表达或功能缺陷时,即可给予诊断。
AS临床诊断共识标准包括均出现表现(几乎全部患者均出现的表现)、经常性表现(超过80%患者出现的表现)以及相关性表现(少于80%患者出现的表现)。均出现的表现包括发育迟滞、语言障碍、运动或平衡障碍,通常是步态障碍和(或)肢体颤动、行为独特包括频繁大笑/微笑;明显的快乐举止、易兴奋性,往往伴有手颤和运动过度行为等。经常性表现包括头围发育延迟、癫痫发作以及特征性异常脑电图(高波幅棘-慢波等)。相关性表现包括平枕/枕骨凹陷、下颌突出、宽嘴、齿缝稀疏、频繁流口水、过多的嘴部动作、肤色及发色浅淡、运动时屈曲手臂、睡眠障碍等。
不同发病机制患者在诊断过程中采取的诊断手段不同。遗传学检测首先选择的是DNA甲基化分析,这类方法包括甲基化多重连接依赖式探针扩增技术(MS-MLPA)、甲基化PCR(MS-PCR)等,可诊断母源15q11.2-q13缺失、父源单亲二体(UPD)或印记缺陷(ID)所致的天使综合征,上述方法可诊断80%左右的患者。如果DNA甲基化分析结果正常,则可以考虑进行*UBE3A*基因序列分析,该方法可使大约10%的患者得到确诊。另外,仍有约10%患者由于尚未明确的致病机制而无法从分子遗传学确诊,只能依据典型表现做出临床诊断。SNP-array芯片分析可以检测15号染色体的微缺失和UPD,但无法从技术上区别天使综合征和普拉德-威利综合征,需结合临床进行诊断,或采用MS-MLPA方法进行验证。
鉴别诊断
AS患者通常存在非特异性精神运动发育迟滞和(或)癫痫,因此鉴别诊断往往具有非特异性。包括脑瘫、非进展性脑病或线粒体脑肌病等,但天使综合征的肢体抖动和不稳定是其与上述疾病的鉴别点。
目前认为,AS需要与如下疾病进行鉴别诊断:
Mowat-Wilson综合征 可表现为喜怒无常、癫痫发作、下颌突出、耳垂突出、言语减退、小脑、便秘,有时还可表现为先天性巨结肠。先天性心脏缺损或胼胝体发育不全也可能发生。Mowat-Wilson综合征通常是由*ZEB2*基因新发突变导致。
Pitt-Hopkins综合征 该病特征是智力障碍、宽嘴和独特的面部特征,以及间歇性过度通气伴呼吸暂停。小头畸形、癫痫发作、共济失调步态和快乐个性等表现与AS有重叠。3岁后患者可有特征性的日间过度换气。该病致病基因为*TCF4*。
Christianson综合征 该病临床特征包括明显快乐的性格、严重的认知延迟、共济失调、小头畸形和癫痫障碍。患者身材瘦小,10岁后可能无法行走,部分患者可能有小脑和脑干萎缩。尽管两者均存在癫痫发作,但脑电图特点有不同表现:AS有特征性的高波幅棘-慢波(1.5~3Hz)特点,而Christianson综合征具有快(10~14Hz)的背景节律。Christianson综合征是由*SLC9A6*基因突变引起的。
Rett综合征 典型表现为出生后早期智力运动发育正常,6~18个月出现认知及运动功能的倒退,语言及社会交往能力下降,头围增长缓慢,丧失已获得的手的精细功能,出现手的刻板动作。出现惊厥、小头畸形和严重言语障碍的女性AS患者需注意与Rett综合征鉴别。Rett综合征的女性患者通常不容易快乐,而患有AS的女性患者则缺乏神经退行性病程和频繁的无目的性的手的动作。在年龄较大女性患者中进行两者的鉴别诊断时存在一定困难,需依靠遗传学检测进行确诊。Rett综合征是由*MECP2*基因突变引起的X连锁隐性遗传病。
以喂养困难和肌张力低下为主要表现的AS患者需注意与普拉德-威利综合征鉴别。此外,其他疾病,如2q23.1微缺失、22q13.3缺失综合征、男性MECP2重复、腺苷酸琥珀酸裂解酶缺乏症、与低甲硫氨酸和高同型半胱氨酸血症相关的亚甲基四氢叶酸还原酶(MTHFR)缺乏症、先天性糖基化障碍、Kleefstra综合征、HERC2相关的认知障碍、WAC相关的智力障碍等亦需酌情鉴别。
治疗
该病迄今尚无特效治疗。主要工作在于诊断后健康管理。目前主要是针对临床表现进行积极地对症及支持治疗,有助于提高AS患儿的生活质量。
当新生儿出现喂养困难时,需要采用特殊奶嘴和其他方法管理患儿吮吸能力弱或不协调的情况;使用抗癫痫药物控制癫痫发作,目前暂无推荐药物;对于出现社会破坏性或自我伤害等不良行为的患儿,建议采用行为疗法进行干预;应在适当的时候尽早使用辅助沟通工具,如图片卡或交流板,以改善患儿言语障碍的情况;利用胸腰椎夹克和(或)手术干预来治疗脊柱侧凸症状。
有一些基因治疗正在积极开展中,比如利用端粒酶抑制剂或反义寡核苷酸激活甲基化沉默的父源UBE3A等位基因,以期达到增加父源*UBE3A*等位基因的表达、补充母源UBE3A等位基因缺陷、最终治疗疾病的目的。
诊疗流程
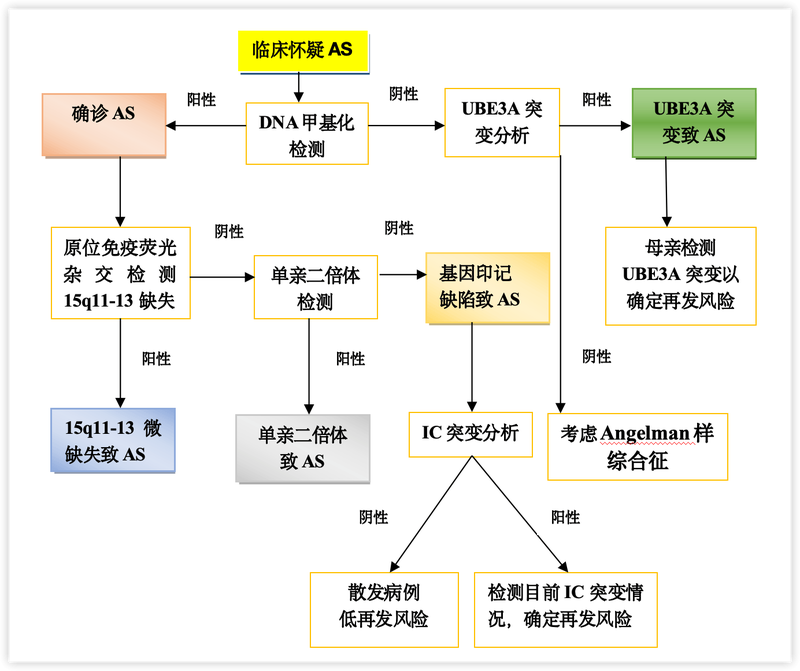
Angelman氏症候群(AS)分子诊疗流程图 AS.Angelman氏症候群(天使综合征);IC.印记中心
参考文献
[1] Jiang Y, Lev-Lehman E, Bressler J, et al. Genetics of Angelman syndrome.Am J Hum Genet,1999,65:1-6.
[2] Clayton-Smith J, Pembrey ME. Angelman syndrome. J Med Genet,1992,29:412-415.
[3] Dagli A, Buiting K, Williams CA. Molecular and clinical aspects of Angelman syndrome. Mol Syndromol,2012,2(3-5):100-112.
[4] Williams CA, Beaudet al, Clayton-Smith J, et al. Angelman syndrome 2005:updated consensus for diagnostic criteria. Am J Med Genet A,2006,140(5):413-418.
[5] Bai JL, Qu YJ, Jin YW, et al. Molecular and clinical characterization of Angelman syndrome in Chinese patients. Clin Genet,2014,85(3):273-277.
[6] https://www.ncbi.nlm.nih.gov/books/NBK1144/
上一篇: 肌萎缩侧索硬化诊疗指南
下一篇: 生物素酶缺乏症诊疗指南




